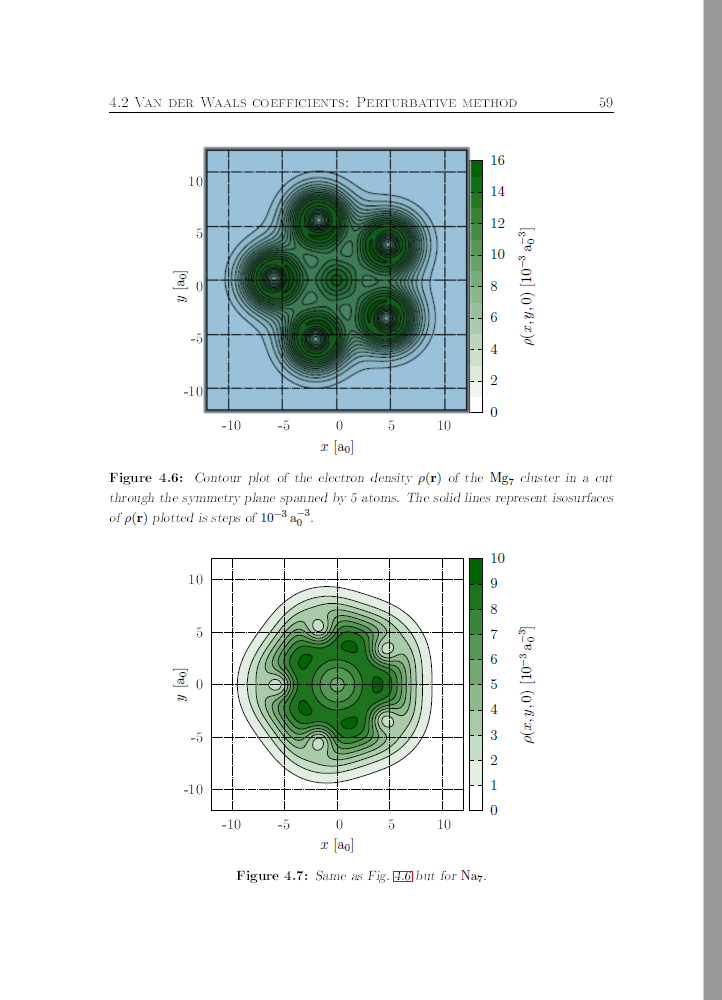
Michael Liebrecht has successfully (and with honors) defended his Ph.D. thesis “Van der Waals potentials between metal clusters and helium atoms obtained with density functional theory and linear response methods”.
Abstract:
The importance of van der Waals interactions in many diverse research fields such as, e. g., polymer science, nano–materials, structural biology, surface science and condensed matter physics created a high demand for efficient and accurate methods that can describe van der Waals interactions from first principles. These methods should be able to deal with large and complex systems to predict functions and properties of materials that are technologically and biologically relevant.
Van der Waals interactions arise due to quantum mechanical correlation effects and finding appropriate models an numerical techniques to describe this type of interaction is still an ongoing challenge in electronic structure and condensed matter theory.
This thesis introduces a new variational approach to obtain intermolecular interaction potentials between clusters and helium atoms by means of density functional theory and linear response methods.
It scales almost linearly with the number of electrons and can therefore be applied to much larger systems than standard quantum chemistry techniques.
The main focus of this work is the development of an ab–inito method to account for London dispersion forces, which are purely attractive and dominate the interaction of non–polar atoms and molecules at large distances.
The first step in that method is to determine the ground states of the two interaction partners with mean-field approaches like density functional theory or Hartree-Fock. Then an iterative scheme that uses linear response functions is employed to calculate the dispersion energy of the two molecules. To obtain meaningful results in reasonable time, it has been necessary to implement the response algorithms so that they do not require any explicit knowledge of unoccupied states. The same response methods are used to construct a very efficient density update scheme that is able to significantly reduce the number of self-consistency iterations of density functional calculations.
It can be shown that our derived equations for the dispersion energy provide the same results as the adiabatic connection dissipation-fluctuation theorem, which can be applied to include correlation effects into density functional calculations. To test our method, the leading van der Waals interaction coefficients for several atom pairs were computed and compared to values from the literature. We found good agreement but also realized that pseudopotentials crucially influence our results.
The construction of intermolecular interaction potentials between clusters and helium atoms requires also the inclusion of the short-ranged Pauli-repulsion. To account for this effect, a semi-empirical helium pseudopotential with a tunable interaction strength is introduced. This pseudopotential is simply added to the effective Hamiltonian of the cluster. To determine the interaction strength of the helium pseudopotential, our results for the Mg-He interaction are fitted to reference potentials found in the literature. It turned out that additional reference potentials for the Na-He and Rb2-He interactions can be approximated remarkably well with the pseudopotential obtained for the Mg-He interaction. Finally, our new approach is applied to construct intermolecular potentials between larger clusters and helium atoms to demonstrate its effectiveness.
Diese Website benutzt Cookies. Wenn du die Website weiter nutzt, gehen wir von deinem Einverständnis aus.